 Significant portions of natural groundwater reserves have been contaminated due to anthropogenic activities that result from by-products of industry, agricultural runoff, and human/animal waste. The contamination at the Department of Energy’s (DOE) Oak Ridge Integrated Field Research Challenge (OR-IFRC) includes radionuclides (e.g., uranium and technetium), nitrate, sulfide, and volatile organic compounds due to nuclear weapon development and processing in the 1940s. A previous study by ENIGMA scientists showed that those contaminants not only affected the quality of groundwater directly but also altered various biogeochemical cycling processes by altering native microbial communities through shot-gun metagenome analysis, resulting in a loss of biodiversity in groundwater communities (Hemme et al., 2010, ISME J 4, 660–672).
Significant portions of natural groundwater reserves have been contaminated due to anthropogenic activities that result from by-products of industry, agricultural runoff, and human/animal waste. The contamination at the Department of Energy’s (DOE) Oak Ridge Integrated Field Research Challenge (OR-IFRC) includes radionuclides (e.g., uranium and technetium), nitrate, sulfide, and volatile organic compounds due to nuclear weapon development and processing in the 1940s. A previous study by ENIGMA scientists showed that those contaminants not only affected the quality of groundwater directly but also altered various biogeochemical cycling processes by altering native microbial communities through shot-gun metagenome analysis, resulting in a loss of biodiversity in groundwater communities (Hemme et al., 2010, ISME J 4, 660–672).
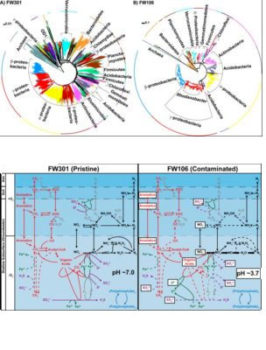
In this study ENIGMA researchers at University of Oklahoma, Lawrence Berkeley National Lab, Montana State University, University of Tennessee and Oak Ridge National Lab further compared the metagenomes from pristine (FW301) and highly contaminated (FW106) wells at this site, aiming to better understand the impact of contaminants on groundwater microbiomes by determining a phylogenetic and metabolic difference between the communities. Proteobacteria (e.g., Burkholderia, Pseudomonas) are the most abundant lineages in the pristine community, though a significant proportion (>55%) of the community is composed of poorly characterized low abundance (individually <1%) lineages. The phylogenetic diversity of the pristine community contributed to a broader diversity of metabolic networks than the contaminated community. Also, the pristine community encodes redundant and mostly complete geochemical cycles distributed over multiple lineages and appears capable of a wide range of metabolic activities. In contrast, many geochemical cycles in the contaminated community appear truncated or minimized due to decreased biodiversity and dominance by Rhodanobacter populations capable of surviving the combination of stresses at the site. These results indicate that the pristine site contains more robust and encodes more functional redundancy than the stressed community, which contributes to more efficient nutrient cycling and adaptability than the stressed community.
More importantly, this study is the first metagenomic characterization of background OR-IFRC groundwater metagenome, and the first formal comparison of metabolic potential, community structure, and gene abundance between background and contaminated OR-IFRC groundwater communities. And the results allow researchers to further test hypotheses and predict the effects of different contamination profiles on groundwater communities and to fine-tune bioremediation strategies of contaminated sites.